Reference
Jian Zhou, Olga G. Troyanskaya. Predicting the Effects of Noncoding Variants with Deep learning-based Sequence Model. Nature Methods (2015).
Code
The code for the standalone version of DeepSEA can be downloaded here (766M). If you need the variant classifiers predictions, you also need to download additional evolutionary conservation score files here (30G).
The code for training the DeepSEA model can be downloaded here. Code + Training data bundle is provided here (3.7G). The genome coordinates of training data can be downloaded here. We now recommend our new library Selene for training purposes.
Update log
03/08/2024: replaced luajit script
05/04/2017: bug fix - Fixed coordinate display error for sequence profiler. If you were previously using the sequence profiler with vcf input, note that the coordinate displayed in the visualization and in the csv file incorrectly started from the variant position (the correct coordinates should start from variant position - 499, and the sequence and predictions displayed were for the correct coordinates).
06/01/2016: minor update - Added chromosome coordinates to sequence profiler outputs. Improvement in help text. Added input file coordinates for download.
10/22/2015: minor update - Added chromatin feature probability difference output.
09/02/2015: minor update - DeepSEA now handles vcf files with more than 5 columns properly. Fixed a bug that causes no output error for some input.
08/26/2015: minor update - we no longer filter out coding variants from input by default.
Input
DeepSEA predicts genomic variant effects on a wide range of chromatin features at the variant position (Transcription factors binding, DNase I hypersensitive sites, and histone marks in multiple hunman cell types. See the full list here.). DeepSEA can also be ultilized for predicting chromatin features for any DNA sequence.
We support three types of input: vcf, fasta, bed. If you want to predict effects of noncoding variants, use vcf format input. If you want to predict chromatin feature probabilities for DNA sequences, use fasta format. If you want to specify sequences from the human reference genome (GRCh37/hg19), you can use bed format. See below for a quick introduction, and we provide detailed description of format requirement on the DeepSEA input page:
Vcf format is used for specifying a genomic variant. A minimal example is
chr1 109817590 - G T(if you want to copy cover this text as input, you need to change spaces to tabs since html webpage can not display tab). The five columns are chromosome, position, name, reference allele, and alternative allele.Fasta format input should include sequences of 1000bp length each. If a sequence is longer than 1000bp, only the center 1000bp will be used. A minimal example is :
>TestSequence TATCTCTCATGTTTCTGGTATAGATGGTATATATGTTAATCTTGTTCCTGAGGTCTGTTTTTTATTTTTGTCATTAAAGT GGGAATTAAATAGTTTTGTAGTGCATATAAATTAAAGAAAAAGTTCACATAAGCATATTTGCCAATCATCTCAAAATGCT ATATTCTCCTTCACGGTTTTGAAAATAATTCAGGGTTTTCTCTTCCTCATTGCTTTCCCACCAACTGACAGTATTATTTT CTTAGTCATTTTACTGACCTTTGAAATTACTCCTTTGAGGTCTTCTAAAAAATTTTATGGGCTCTGCTGCTTTTTGGTGG CCTCCTTGTATCATTTATTCTATTACAGGACGACTTACAAAAGGAAGCACATAAATTGACCCATATACATATCCTATCAT TGGGGAGTTTCTGTGCAAATGTTATTTATTGGAAGCTATTACTAAGAATTGTAAGAAAAATAATTGGTATTGATGCAGCT AGTATGGTTCCTGTAATTATCGTACTCAGCCACGTAAATCATAGCTATATGTAGCCAAAGATCCATGAACAAAATTTCCA GTAACATCATTATAATTCAAAAGGCAGACTTTCAGAACCAGACAGACTTGAATTTAAATTCTAGCTTTACCACACATGAA TTTAACCTTGTGGAAGGTTAACCTATCTAAACTCATGTTTCTTCATTGGTAGCTGATAAAATTAAGGATCATGTATATAA CCACCTAGTAGAGTTGTTTAAGAAACTGTTAGAATTCCATAAATTGTTAGTATTAATGAGTTTTTGTTGGACATGTGTTA GGCTAGGCCACTCCTTGACCTTCATAGAGGTATGGATTATGACACAAATTCTAAACTGTAGGTAGGCATGGCTTTGTAGC AAGTATTAAAATAGTAAATATTTTATTTTTATAAGATAAATGTAAACCTTTTAAAAGTTTCATTACATTTGTATTTATGA AATATCATCCTATATCAACTATAGAGAGAAGATCGCAAGA- Bed format provides another way to specify sequences in human reference genome (hg19). The bed input should specify 1000bp-length regions. A minimal example is
chr1 109817091 109818090. The three columns are chromosome, start position, and end position.
We recommend using the server if you have <50,000 variants or sequences. For larger set, you may run the standalone version on your local machine.
We support only GRCh37/hg19 genome coordinates. You can use LiftOver to convert your coordinates to the correct version.
Output
You will be given a download link to a zip file including all the output files. (Note that the order of rows in output can be different from the input because we sort all the regions and variants!)
For sequences input in fasta/bed format, the output will be a single tab-delimited file providing 919 chromatin feature probability predictions for each of the input sequences.
For variants input in vcf format, the output will include six tab-delimited files:
- infile.vcf.out.ref: Chromatin feature probabilities for sequences carrying the reference allele.
- infile.vcf.out.alt: Chromatin feature probabilities for sequences carrying the alternative allele.
- infile.vcf.out.logfoldchange: Chromatin feature probability log fold changes
log(p_alt/(1-p_alt))-log(p_ref/(1-p_ref))for each variant. Computed by comparing the 'ref' and 'alt' files. Note that log fold change can be noisy when probabilities are small, therefore this score should be considered together with probability differences and E-values when evaluating the impact of a variant. - infile.vcf.out.diff: Chromatin feature probability differences
p_alt - p_reffor each variant. Computed by comparing the 'ref' and 'alt' files. This is a robust measure for variant effects. - infile.vcf.out.evalue: E-values for the chromatin feature effects.
- infile.vcf.out.funsig: Functional significance score for each variant.
- infile.vcf.out.snpclass: The probability of a SNP being a eQTL or trait-associated (GWAS) SNPs given by functional variant prioritization classifiers (Note that only common variants were used in training these classifiers, thus be careful on interpreting the predictions on rare variants).
An example infile.vcf.out.logfoldchange output format for variant input (vcf) (other output files are similarly organized:
| chr | pos | name | ref | alt | 8988T|DNase|None | AoSMC|DNase|None | ... |
|---|---|---|---|---|---|---|---|
| chr1 | 109817590 | - | G | T | 3.89E-01 | 2.34E+00 | ... |
| chr10 | 23508363 | - | A | G | 1.56E-01 | 8.59E-02 | ... |
| chr16 | 52599188 | - | C | T | -7.13E-02 | 2.65E-02 | ... |
| chr16 | 209709 | - | T | C | 7.28E-02 | 3.60E-01 | ... |
Chromatin features such as "8988T|DNase|None" are named by Cell Type_Chromatin Feature Type_Treatment convention. The values shown in this example are log fold changes.
We also provide visualization of the results (see below screenshot, or you can checkout an example output here).
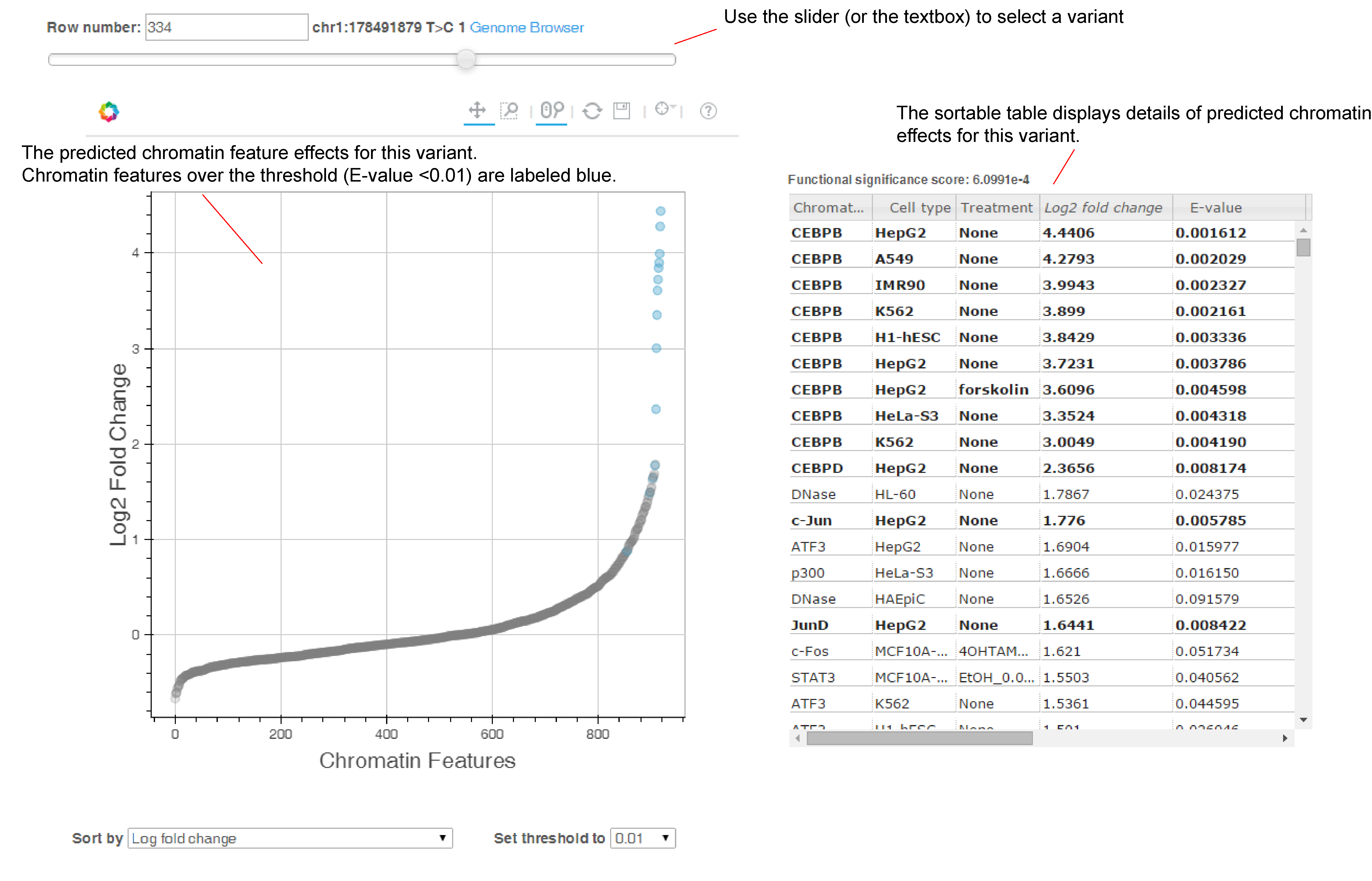
Use the slider to select the variant/sequence to show. Hover over a data point to show the details of this chromatin feature (Chromatin feature name, Cell type, Treatment, and Probability/Log Fold Change). By default we sort the chromatin features by log probability fold change for variants, and by probability for sequences, and you can switch to sorting by chromatin feature class, chromatin feature name, or cell types.
Note we will show the visualization of the top 500 variants/sequences if the input includes more than 500 variants/sequences.
Sequence Profiler
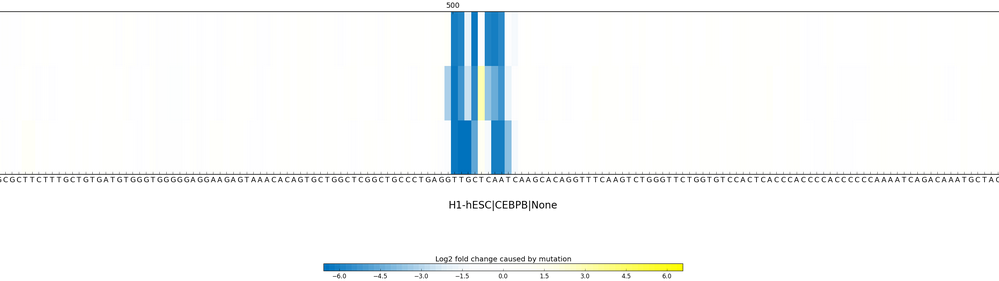
Sequence Profiler performs "in silico saturated mutagenesis" analysis for discovering informative sequence features within any sequence. Specifically, it performs computational mutation scanning to assess effect of mutating every base of the input sequence on chromatin feature predictions. This method for context-specific sequence feature extraction fully utilizes the DeepSEA’s capability of utilizing flanking context sequences information.
We support three types of input for specifying a sequence to analyze: vcf, fasta, bed. See the Input section for a brief introduction to the formats. Note that sequence profiler only accepts one sequence / region / variant as input. If a variant is given, we perform analysis on the sequence carrying the alternative allele (if you want to analyze the reference allele, just use bed or fasta format input instead).
You also need to specify the chromatin feature that you want to analyze (e.g. CEBPB in H1-hESC cell with no treatment). If you have no idea which chromatin feature to look at, you may provide your input to DeepSEA and check which chromatin features are predicted to be on for your sequence.
Sequence Profiler outputs effects for each of three possible substitutions of all 1000 bases on the chromatin feature you specified. See an example output here.
Q&A
What should I use for prioritizing variants? We provide three classifiers trained on HGMD mutations, eQTLs and GWAS lead SNPs respectively. The first is likely appropriate for de novo mutation while the later two are likely appropriate for common variants. We also provide an unsupervised score named functional signficance score - it is not biased by any variant training set but may not have as strong performance as classifiers. We highly recommend utilizing the predictions for individual chromatin features. Depending onthe specific goal you have, the classifiers we provide here may not suit your need. If so, consider training a regularized logistic regression model for your need using DeepSEA chromatin effect predictions (e.g. differences in probability). This can be done with R packages such as xgboost or glmnet. You may want choose appropriate positives (e.g. annotated pathogenic noncoding mutation, or common variants with functional evidences), and appropriate negatives (e.g. common variants from healthy individual, rare variant from individual, or spatially adjacent variants close to a annotated variants) depending on the set of variants you want to prioritize.
What is functional significance score? Functional significance score is a measure of the signficance of magnitude of predicted chromatin effect and evolutionary conservation. Specifically functional significance score is computed as the product of the geometric mean E-value across chromatin features and the geometric mean E-value of the evolutionary conservation scores. Functional significant score is meant to be a general functionality score not specific to a particular purpose,thus we use it in ranking variants for display. Please note that functional significance score is not P-value and should not be used as P-value.
Can DeepSEA predict effects of INDELs? Yes. We support both single nucleotide substitution and small insertion/deletions (up to 100bp). For InDels, the reference string (forth column) must include the base before the insertion/deletion event, and the base must match the reference genome at the genomic position specified by the first and second columns. For example,
19 41304596 rs10680577 T TTACTspecifies a small insertion of "TACT".What is the E-value of a variant for a chromatin feature? E is short for 'Expect', and E-value is defined as the expected proportion of SNPs with larger predicted effect (from reference allele to alternative allele) for this chromatin feature. The predicted effect magnitude is measured as the product of relative and absoluted change, i.e.
|log(p_ref/(1-p_ref))-log(p_alt/(1-p_alt))| * |p_ref-p_alt|. E-value is computed based on the empirical distributions of predicted effects for 1000 Genomes SNPs.What is the interpretation of the predicted probabilities for chromatin features of a sequence / genomic region? The probability output of DeepSEA for a chromatin feature is the probability of observing a binding event (ChIP peak) at the center 200bp region of the sequence, with the assumption that the overall frequency of binding events is the same as the training data of DeepSEA. For variant input (vcf format), we compute the probabilities for both the reference allele and the alternative allele for each variant.
How accurate are DeepSEA predictions for a specific chromatin feature? See AUCs here. AUC can be interpreted as the probability of ranking a random positive example higher than a random negative example. The median AUC for TF, DNase I hypersensitive sites, and histone marks are 0.958, 0.923, 0.856 respectively. For details of AUC calculation please refer to our manuscript.
What is normalized probability vs. probability for chromatin feature predictions? The overall scale of predicted probabilities for a chromatin feature depends on the proportion of positive examples in the DeepSEA training set (uniformly processed peaks from ENCODE and Roadmap Epigenomics projects). The proportion of positive examples can be affected by many experiment specific factors. Normalized probabilities removed the effect of positive proportions in training data on the scale of predicted probabilities by assuming uniform positive proportion(5%) across all chromatin features. Specifically, we used the formula
1/(1+exp(-( log(P/(1-P))+log(5%/(1-5%))-log(c_train/(1-c_train ))) ))where P is the probability given by the DeepSEA model, and c_train is the proportion of positive examples for this chromatin feature in the training data. The proportion of positive examples for all the chromatin features in DeepSEA training data can be downloaded here.
For Sequence Profiler, does no effect detected for mutating a base suggest that base is not predictive for the chromatin feature? Be aware of the redundancy of binding sites which is quite common. If multiple redundant binding sites exist, it may buffer the effect of mutating a base on one of the site. If you suspect this is the case, try to mutate multiple copies of the sequence elements at the same time. You can do this by generating sequence of your design in fasta format and analyze output of DeepSEA.